Automatische Übersetzung anzeigen
Dies ist eine automatisch generierte Übersetzung. Wenn Sie auf den englischen Originaltext zugreifen möchten, klicken Sie hier
#Neues aus der Industrie
{{{sourceTextContent.title}}}
Der Blinddarm kann sich nicht nur entzünden, sondern sich auch krebsartig verändern! Er kann ein lebensbedrohliches Risiko darstellen, ähnlich wie Darmkrebs.
{{{sourceTextContent.subTitle}}}
Der Blinddarm kann sich nicht nur entzünden, sondern sich auch krebsartig verändern! Er kann ein lebensbedrohliches Risiko darstellen, ähnlich wie Darmkrebs.
{{{sourceTextContent.description}}}
01
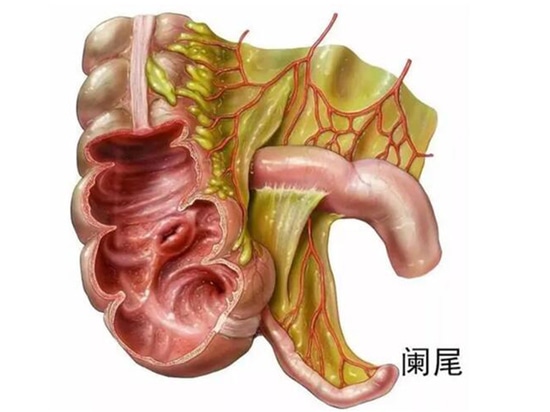
Anatomie und Funktion des Blinddarms
Der Blinddarm ist ein intraperitoneales Organ, und das Peritoneum, das ihn umhüllt, bildet entlang einer Seite seiner Wand ein doppellagiges dreieckiges Mesenterium, das so genannte Blinddarm-Mesenterium. Daher ist der Blinddarm ein frei bewegliches Organ in der Bauchhöhle. Da das Mesenterium oft kürzer ist als der Wurmfortsatz, befindet sich der Blinddarm in der Regel in einem gewundenen Zustand.
Der Blinddarm hat ein regenwurmähnliches Aussehen mit einer Länge von etwa 5 bis 10 cm und einem Durchmesser von etwa 0,5 cm. Seine Gewebestruktur ähnelt der des Dickdarms und besteht von innen nach außen aus vier Schichten: Schleimhautschicht, Submucosaschicht, Muskelschicht und Serosaschicht. Die Schleimhaut- und die Submukosaschicht des Wurmfortsatzes enthalten reichlich lymphatisches Gewebe, so dass der Wurmfortsatz ein lymphatisches Organ mit bestimmten Immunfunktionen ist.
Untersuchungen deuten darauf hin, dass der Blinddarm an der Produktion und Reifung von B-Lymphozyten beteiligt ist. Das lymphatische Gewebe im Blinddarm beginnt nach der Geburt zu entstehen, erreicht seinen Höhepunkt im Teenageralter, nimmt allmählich ab und verschwindet nach dem 60. Die Entfernung des Wurmfortsatzes bei Erwachsenen hat daher keine wesentlichen Auswirkungen auf die Immunfunktion des Körpers.
02
Blinddarmtumore
Die Blinddarmentzündung ist eine häufige Erkrankung des Wurmfortsatzes, die durch entzündliche Veränderungen aufgrund verschiedener Faktoren gekennzeichnet ist. Es handelt sich um eine häufige chirurgische Erkrankung, die am häufigsten bei jungen Menschen auftritt und bei Männern häufiger vorkommt als bei Frauen. Die akute Appendizitis ist klinisch häufiger und betrifft Menschen jeden Alters, auch Schwangere. Die chronische Blinddarmentzündung ist weniger häufig.
Blinddarmtumore/Krebs: Obwohl die Inzidenz von Blinddarmtumoren gering ist, ist in der Bevölkerung eine steigende Tendenz bei Krebserkrankungen des Verdauungssystems zu beobachten[1]. Als Mitglied der Tumorfamilie stellen Blinddarmtumoren trotz ihrer scheinbar unbedeutenden Größe ein ähnliches lebensbedrohliches Risiko dar wie Darmkrebs.
Allgemein lassen sich Blinddarmtumore in Tumore epithelialen Ursprungs (wie Adenome oder Adenokarzinome) und Tumore nicht epithelialen Ursprungs (wie neuroendokrine Tumore oder Lymphome) einteilen. Epitheliale Blinddarmtumoren werden auf der Grundlage der Schleimproduktion in zwei verschiedene Klassen eingeteilt, die sich in ihrem biologischen Verhalten und ihrer Prognose deutlich unterscheiden. Die Weltgesundheitsorganisation (WHO) bezeichnet die meisten nicht invasiven epithelialen Tumoren als niedriggradige muzinöse Blinddarmneoplasien (LAMN), bei denen es sich um gut differenzierte Adenome handelt, die in der Lage sind, außerhalb des Blinddarms bösartig zu wachsen, was zu einer extrazellulären Muzinansammlung führt. Diese Muzine können in zellulärer oder nicht-zellulärer Form vorliegen, und diese Tumoren haben ein potenziell bösartiges biologisches Verhalten, wie z. B. die Verursachung einer Blinddarmperforation, einer Fibrose der Blinddarmwand, einer Muzinbildung in der Blinddarmwand und einer extrazellulären Muzinansammlung in den umgebenden Weichteilen. Hochgradige muzinöse Blinddarmneoplasien (HAMN) weisen im Vergleich zu LAMN eine aggressivere Zellatypie auf.
Die häufigste Art von Tumoren nicht-epithelialen Ursprungs im Blinddarm sind neuroendokrine Tumoren (NETs). Histologisch ähneln sie NETs in anderen Teilen des Verdauungssystems und sind häufig asymptomatisch, da sie in der Regel zufällig nach einer Appendektomie entdeckt werden. Zu den anderen seltenen Tumoren nicht-epithelialen Ursprungs im Blinddarm gehören gastrointestinale Stromatumoren und Lymphome. Die meisten Appendix-NETs haben ein niedrigeres Tumorstadium, was zu einer relativ guten Prognose führt. Die 5-Jahres-Überlebensrate für Patienten mit lokalisierten Läsionen liegt bei 95 % bis 100 %. Obwohl die Tumorgröße mit der Überlebensrate korreliert, zeigen Untersuchungen, dass sich die 5-Jahres-Gesamtüberlebensrate zwischen Tumoren, die kleiner als 1 cm sind, und solchen, die zwischen 1 cm und 2 cm groß sind, nicht wesentlich unterscheidet. Die 5-Jahres-Überlebensrate für Tumore, die größer als 2 cm sind, liegt bei 70,5 %. Patienten mit Fernmetastasen haben eine 5-Jahres-Überlebensrate von weniger als 25 % mit einer mittleren Überlebenszeit von etwa 31 Monaten.
03
Marker für Blinddarmtumore
Adenokarzinom des Wurmfortsatzes: Dieses lässt sich in muzinöse und nicht-muzinöse Typen einteilen, wobei das muzinöse Adenokarzinom eine hochgradige zelluläre Atypie aufweist und über 50 % der Läsionen extrazelluläres Muzin enthalten[2]. Das Adenokarzinom des Blinddarms weist typischerweise p53, CD44 und CDX2 auf.
Muzinöse Tumore des Blinddarms: Die Serumtumormarker CEA, CA19-9 und CA125 können für die Diagnose und die Beurteilung des Krankheitsstatus von Schleimhauttumoren des Blinddarms verwendet werden. Mutationen in den Genen KRAS, TP53 und SMAD4 können zwischen HAMNs und LAMNs unterscheiden, obwohl sie nach Ansicht von Experten keine Bedeutung für die Diagnose oder Behandlung haben.
04
Behandlung von Blinddarmtumoren
Derzeit gibt es keinen einheitlichen Konsens über die Behandlung von Blinddarmtumoren. Experten schlagen vor, dass die Behandlung von Blinddarmtumoren auf dem Gesamtzustand des Patienten, dem histopathologischen Typ des Tumors, dem Ausmaß der Beteiligung und den Entwicklungstendenzen basieren sollte, wobei die Operation die primäre Behandlungsmethode ist[3]
05
Genetische Anfälligkeit von Blinddarmtumoren[4]
Diese Studie umfasste Patienten mit Blinddarmkrebs, die sich vom 1. März 2012 bis zum 31. Dezember 2016 in einem klinischen Testlabor einem Keimbahnmutationstest für 14 Krebsanfälligkeitsgene unterzogen. Die klinische Anamnese, die persönliche Anamnese und die Familienanamnese der Patienten wurden aus den von den klinischen Ärzten ausgefüllten Untersuchungsanforderungsformularen erhoben. Die Multi-Gen-Tests wurden mittels gezielter individueller Erfassung, Sequenzierung und Analyse chromosomaler Rearrangements durchgeführt. Die primären Endpunkte waren Keimbahnmutationen, Inzidenz und Spektrum bei Patienten mit Blinddarmkrebs. Keimbahnmutationen in den Genen APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11 und TP53 wurden mit einer Anfälligkeit für Magen-Darm-Krebs in Verbindung gebracht.
In der Kohorte von 131 Patienten mit Blinddarmkrebs (90 Frauen [68,7 %]) wurden bei 16 Patienten (15,11 %) 5 schädliche Sequenzvarianten festgestellt. Auch bei der Beschränkung auf 74 Patienten mit Blinddarmkrebs als erstem und einzigem Primärtumor wiesen 8 Patienten (10,8 %) mindestens eine schädliche Sequenzvariante in Krebsanfälligkeitsgenen auf. Insgesamt wurden bei 6 Patienten (4,6 %) schädliche Sequenzvarianten in MUTYH beobachtet (5 Fälle mit einem Allel MUTYH, 1 Fall mit einem Doppelallel MUTYH). Vier Patienten mit Lynch-Syndrom (3,1 %) wiesen alle MLH1-Gensequenzvarianten auf, wobei drei Fälle im Alter von 50 Jahren oder darüber diagnostiziert wurden. Drei Patienten (8,1 %) hatten schädliche Sequenzvarianten in anderen Krebsanfälligkeitsgenen (3920 Fälle in APC [c.1307T>A, p.I2K], 2 Fälle in CHEK470 [c.157T>C, p.I1T], 4 Fälle in SMAD263 [c.287 98dup, p.L14IFS*1], 53 Fälle in TP524 [c.175G>A, p.R<>H]).
Schlussfolgerung: Von 10 Blinddarmkrebspatienten, die sich einer erblichen Krebsanfälligkeitsprüfung unterziehen, trägt eine Person genetische Mutationen, die mit einer Krebsanfälligkeit assoziiert sind. Angesichts der hohen Häufigkeit und des breiten Spektrums genetischer Keimbahnvariationen legen diese Daten nahe, dass bei allen Patienten, bei denen diese seltene bösartige Erkrankung diagnostiziert wird, eine genetische Untersuchung erforderlich sein könnte.
Referenzen:
[1] Journal of Gastrointestinal Surgery, 2015, 19(4): 743-750.
[2] 4th ed. Lyon, Frankreich: IARC Press, 2010.
[3] Chinese Expert Consensus on Multidisciplinary Comprehensive Treatment of Appendiceal Tumors (Ausgabe 2021).
[4] JAMA Oncol. 2022 Nov 11;9(1):95-101.
Erklärung: Dieser Artikel ist nur zur Weitergabe bestimmt. Sollte es Probleme mit dem Urheberrecht geben, kontaktieren Sie uns bitte so schnell wie möglich, und wir werden dies so schnell wie möglich korrigieren. Wir danken Ihnen!