Automatische Übersetzung anzeigen
Dies ist eine automatisch generierte Übersetzung. Wenn Sie auf den englischen Originaltext zugreifen möchten, klicken Sie hier
#Neues aus der Industrie
{{{sourceTextContent.title}}}
Präsentation von Tumor-Neoantigenen
{{{sourceTextContent.subTitle}}}
Präsentation von Tumor-Neoantigenen
{{{sourceTextContent.description}}}
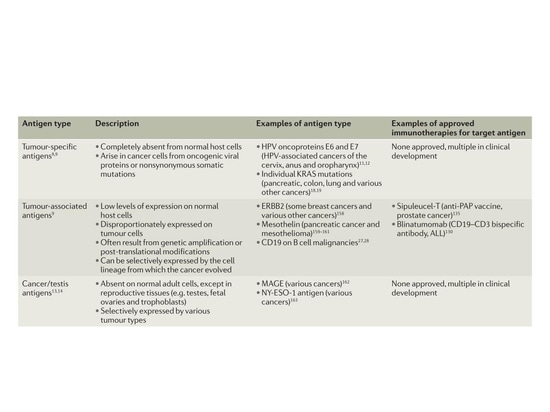
Klassifizierung von Tumorantigenen
Die jüngsten Fortschritte bei der Genomsequenzierung zeigen, dass Krebs während der Entstehung und Entwicklung Zehntausende verschiedener somatischer Zellmutationen aufweist. Die meisten dieser Mutationen bringen keine inhärenten Wachstumsvorteile mit sich (Passagiermutationen) und sind häufig das Ergebnis einer genomischen Instabilität innerhalb des Tumors.
Eine kleine Gruppe von Krebsmutationen stört die normale zelluläre Regulation und trägt dazu bei, das Krebswachstum voranzutreiben und die Resistenz gegen gezielte Therapien zu erhöhen (Treibermutationen). Bislang wurden etwa 140 Gene identifiziert, die die Tumorentstehung fördern können. Sowohl Treiber- als auch Passagiermutationen können jedoch die kodierende Aminosäuresequenz verändern, was als synonyme Mutationen bezeichnet wird und zu mutierten Proteinen führt, die in normalen Zellen nicht exprimiert werden. Diese abweichenden Proteinsequenzen werden zu kurzen Peptiden verarbeitet und binden an den Haupthistokompatibilitätskomplex (MHC; beim Menschen auch als humanes Leukozytenantigen HLA bekannt), wodurch sie von T-Zellen als fremde Antigene erkannt werden [1].
Aufgrund ihrer selektiven Expression in Tumoren werden tumorspezifische Antigene (TSAs), die durch synonyme Mutationen und andere genetische Veränderungen entstehen, als Neoantigene bezeichnet. In menschlichen Tumoruntergruppen mit viraler Ätiologie, wie dem Merkelzellkarzinom (MCC), das mit dem Merkelzell-Polyomavirus (MCPyV) assoziiert ist, und Gebärmutterhals-, Mundhöhlen- und anderen ortsspezifischen Krebsarten, die mit dem humanen Papillomavirus (HPV) assoziiert sind, stellen Proteine, die vom viralen offenen Leseraster kodiert werden, eine weitere Art von Neoantigen dar. Neben den TSAs gibt es zwei Hauptklassen von Tumorantigenen. Tumor-assoziierte Antigene (TAAs) werden in bösartigen Zellen übermäßig häufig, aber auch in geringen Mengen in normalen Zellen exprimiert. Krebs-/Hoden-Antigene (CTAs) werden von verschiedenen Tumorarten und reproduktiven Geweben (z. B. Hoden, fötale Eierstöcke und Nährböden) exprimiert, sind aber in anderen normalen erwachsenen Geweben nur begrenzt vorhanden und fehlen in der Regel in normalen reproduktiven Zellen, da diese Gewebe keine MHC-Klassenmoleküle exprimieren. Neoantigene können auf genomischer Ebene durch einzelne Nukleotidvariationen (SNVs), Baseninsertionen und Genfusionen, auf transkriptioneller Ebene durch selektives Spleißen, Polyadenylierung (pA), RNA-Editierung und so genannte nicht-kodierende Regionen sowie auf Proteinebene durch gestörte Translation und posttranslationale Modifikationen entstehen.
▲Klassifizierung von Tumorantigenen[1]
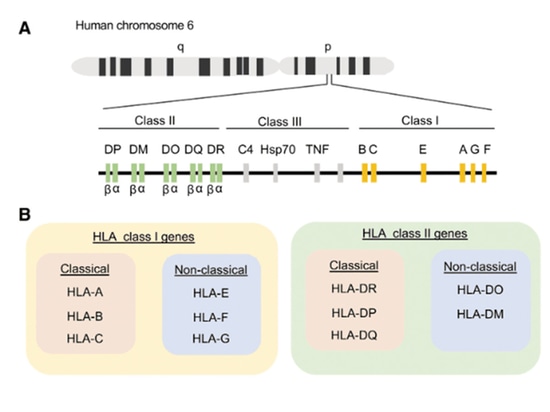
Klassifizierung von HLA
Die Aktivierung von T-Zellen hängt von der gleichzeitigen Erkennung von fremden Peptidfragmenten und eigenen MHC-Molekülen ab, ein Phänomen, das als MHC-Restriktion bekannt ist. CD8+ T-Zellen werden durch MHC-I eingeschränkt, während CD4+ T-Zellen durch MHC-II eingeschränkt werden. Der Haupthistokompatibilitätskomplex (MHC), der beim Menschen als humanes Leukozytenantigen (HLA) bezeichnet wird, ist auf Chromosom 6 des menschlichen Genoms kodiert. Es handelt sich um einen hochpolymorphen Genkomplex, der für Zelloberflächenmoleküle kodiert, die auf die Präsentation und Erkennung von eigenen und fremden Peptiden spezialisiert sind. HLA wird aufgrund seiner Funktion und Struktur in drei Klassen eingeteilt: HLA-I, HLA-II und HLA-III. Moleküle der HLA-Klasse werden auf der Oberfläche von kernhaltigen Zellen exprimiert, mit Ausnahme von Keimzellen und einigen neuronalen Zellen. Im Gegensatz zu den Molekülen der Klasse HLA-I befinden sich die Moleküle der Klasse HLA-II typischerweise auf professionellen Antigen-präsentierenden Zellen (APCs) wie B-Zellen, Makrophagen, dendritischen Zellen, Langerhans-Zellen, Thymusepithel und aktivierten (und nicht ruhenden) T-Zellen. Struktur und Funktion der HLA-III-Klassenmoleküle sind nicht genau bekannt, aber es ist bekannt, dass sie am Entzündungsprozess beteiligt sind, ohne direkt an Antigene zu binden.
HLA wird weiter in klassische und nicht-klassische Gene unterteilt. Zu den klassischen HLA-I-Genen gehören HLA-A, HLA-B und HLA-C, während zu den nicht-klassischen Allelen HLA-E, HLA-F und HLA-G gehören. Zu den klassischen HLA-II-Genen gehören HLA-DR, HLA-DP und HLA-DQ, während zu den nichtklassischen Allelen HLA-DO und HLA-DM gehören. Menschliche CD8+ T-Zellen erkennen Peptide, die von den klassischen HLA-A und HLA-B und in geringerem Maße auch von HLA-C präsentiert werden. Menschliche CD4+ T-Zellen erkennen Peptide, die von HLA-DR, HLA-DQ und HLA-DP präsentiert werden. HLA-I besteht aus drei extrazellulären Domänen (α1, α2 und α3), die nicht kovalent an ein β2-Mikroglobulinmolekül gebunden sind. HLA-II ist ein Heterodimer, das aus einer α- und einer β-Kette besteht. Die extrazellulären Domänen von HLA bilden einen Antigen-bindenden Spalt, der aus zwei α-Helices besteht, die antiparallele β-gefaltete Blätter umgeben. Dadurch wird eine Plattform geschaffen, die ein kurzes Aminosäuresegment (aa), ein so genanntes Peptid, aufnehmen kann. Diese Peptide binden sich an den Boden der Bindungsfurche durch Wechselwirkungen mit spezifischen Aminosäuren (sogenannte Ankerreste). Aufgrund der geschlossenen Struktur der HLA-I-Bindungsfurche werden in der Regel kleine Peptide mit 8-10 Aminosäuren gebunden, während HLA-II längere Peptide mit mehr als 11 Aminosäuren binden kann.
▲Klassifizierung diffuser Gliome bei Erwachsenen[2]
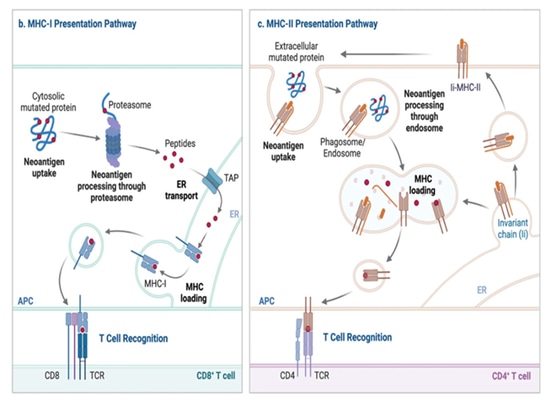
Präsentation von Neoantigenen
Die Präsentation von MHC-I-Proteinen geht in erster Linie auf Peptide im Inneren der Zelle zurück. Während der Homöostase werden zelluläre Proteine durch Proteasomen abgebaut, wobei kleine Peptide entstehen. Proteasomen sind Multiproteinkomplexe, die Proteine in kleine Peptidfragmente zerlegen. Während einer Virusinfektion induziert die Wirkung von Interferonen die Bildung eines alternativen Proteasomkomplexes, des Immunoproteasoms, der die Bildung von MHC-I-präsentierten Peptiden verstärkt. Folglich werden virale Proteine bei ihrer Synthese durch das Immunoproteasom abgebaut, entweder als vollständig gefaltete Proteine oder als defekte ribosomale Produkte. Dies führt zur Produktion von kleinen Peptidfragmenten, die aus dem infizierenden Virus stammen und durch zytoplasmatische Aminopeptidasen weiter modifiziert werden können.
Diese Peptidfragmente werden dann durch einen Proteintransportmechanismus in das endoplasmatische Retikulum (ER) transportiert, der als Antigen Processing Related Transport Protein (TAP) bekannt ist. Im ER werden die Peptide durch die Aminopeptidase 1 des endoplasmatischen Retikulums (ERAP1) weiter modifiziert, wenn sie nach der Verlagerung durch das TAP zu lang für die MHC-I-Bindung sind. Im ER assoziieren leere MHC-I-Moleküle mit Peptidladekomplexen (PLC), zu denen Chaperonproteine wie Tapasin und Calnexin gehören. Der PLC hält das leere MHC-I-Molekül in einer für Peptide empfänglichen Konformation, und wenn Peptide durch den TAP transloziert werden, erleichtert diese Konformation die Peptidbindung. TAP ist ebenfalls ein Teil von PLC. Durch die Peptidbindung wird das MHC-I-Protein stabilisiert und von den Qualitätskontrollpartnern des endoplasmatischen Retikulums befreit, so dass es über den Golgi-Apparat zur Zelloberfläche transportiert wird.
Dieser Prozess ermöglicht es CD8+ T-Zellen, im zellulären Proteinrepertoire nach Anzeichen für eine Infektion oder Malignität zu suchen. In bestimmten Untergruppen dendritischer Zellen gibt es jedoch einen alternativen Weg, die so genannte Cross-Präsentation, bei der extrazelluläre Antigene aufgenommen, retrograd von Phagosomen in das Zytoplasma transportiert und anschließend in Proteasomen und dem ER für die MHC-I-Beladung abgebaut werden.
▲Neoantigen-Produktion und Präsentationswege[3]
Beim Menschen gibt es über 24 000 verschiedene Allele der menschlichen Leukozytenantigenklasse I (HLA-I, einschließlich HLA-A, -B und -C) und der Klasse II (HLA-DR, HLA-DQ und HLA-DP), was zu einem vielfältigen Polymorphismus führt. Die Kombination dieser Allele trägt zur Vielfalt des Polymorphismus bei. Die HLA-Allele des Patienten bestimmen sein tumorspezifisches Neoantigen-Repertoire, das von T-Zellen erkannt werden kann. Darüber hinaus beeinträchtigt der HLA-Verlust der Heterozygotie (HLA-LOH), der bei 40 % der nicht-kleinzelligen Lungenkarzinome auftritt, die Neoantigenpräsentation und begünstigt so die Umgehung des Immunsystems. Daher ist einer der entscheidenden ersten Schritte bei der Neoantigen-Vorhersage die Identifizierung des HLA-Genotyps des Patienten. Um dieses Ziel zu erreichen, können nun verschiedene Berechnungsmethoden auf Next-Generation-Sequencing-Daten (NGS) angewendet werden.
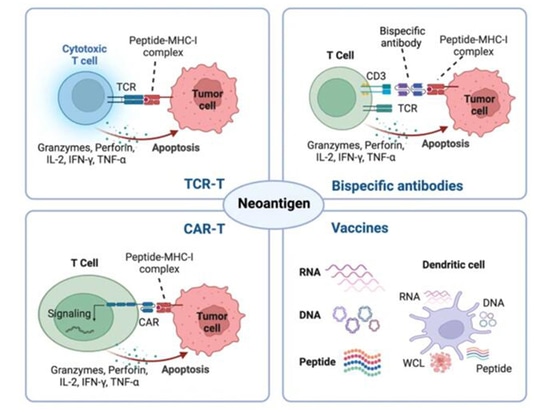
Therapeutische Strategien auf der Grundlage von Neoantigenen
Da es keine thymische Selektion und keine zentrale Toleranz gibt, lösen tumorspezifische Neoantigene, die durch genetische Veränderungen entstehen, hochaffine T-Zellen aus. Unter Ausnutzung ihrer Tumorspezifität und Immunogenität dienen Neoantigene als neue Ziele für die Krebsimmuntherapie, darunter Tumorimpfstoffe, adoptive Zelltherapien (ACT), antikörperbasierte Behandlungen und potenzielle Prädiktoren für Immun-Checkpoint-Inhibitoren (ICBs) [3].
▲Molekulare Merkmale des diffusen hochgradigen Glioms bei Kindern[3]
Das neue Antigen setzt sich aus personalisierten neuen Antigenen zusammen, die spezifisch auf jeden Patienten ausgerichtet sind, oder aus gemeinsamen neuen Antigenen, die bei vielen Krebspatienten exprimiert werden. Therapien, die auf gemeinsamen neuen Antigenen basieren, sind ressourcen- und zeiteffizienter als personalisierte neue Antigentherapien. Da personalisierte neue Antigene patientenspezifisch sind, können sie nicht für die Behandlung einer großen Zahl von Patienten eingesetzt werden. Mit den jüngsten Fortschritten in der Hochdurchsatz-Sequenzierung ermöglichen personalisierte neue Antigene dem Immunsystem, immunogene Epitope auf bösartigen Tumoren ohne vordefinierte gemeinsame Antigene anzuvisieren.
Beziehung zwischen TMB und TNB
Bei den meisten Melanompatienten ist die TMB größer als 10, was zur Bildung wirksamerer Neoantigene führt. Auf der Grundlage dieser Daten lässt sich vorhersagen, dass bei einem TMB-Wert von mehr als 10 im Tumor eine beträchtliche Anzahl von Neoantigenen gebildet werden wird. Liegt die TMB zwischen 1 und 10, besteht immer noch die Möglichkeit, Neoantigene zu tragen. Liegt die TMB im Tumor unter 1, ist es im Allgemeinen schwierig, Neoantigene zu erzeugen, die von T-Zellen erkannt werden können. Tumore mit einem hohen TMB-Wert (>10) deuten darauf hin, dass sich mehr Tumor-Neoantigene auf der Oberfläche der Tumorzellen befinden, was zu einer effektiveren Abtötung durch Immunzellen führt. Außerdem sprechen Patienten mit einem hohen TMB-Wert möglicherweise besser auf die Behandlung mit Immun-Checkpoint-Inhibitoren an.
tumor-TMB und das Potenzial zur Bildung von Neoantigenen[4]
Die Herausforderungen der klinischen Anwendung
1. Immuntherapien, die auf neuen Antigenen beruhen, zeigen nur bei einer kleinen Teilmenge der gut dokumentierten Patientenreaktionen objektive Wirksamkeit. Daher sind erhebliche Verbesserungen erforderlich, um die klinischen Ergebnisse zu verbessern. Dazu gehören eine höhere Genauigkeit bei der Vorhersage neuer Antigene, die Überwindung der Immunumgehung und die Optimierung der Pipeline für den Produktionsprozess.
2. Begrenzte Genauigkeit bei der Vorhersage neuer Antigene: Die breite Anwendung der personalisierten Immuntherapie wird durch die begrenzte Entdeckung krebsspezifischer neuer Antigene eingeschränkt, was auf die Heterogenität der Mutationslast und die erheblichen Unterschiede in der Präsentation neuer Antigene bei den verschiedenen Tumortypen zurückzuführen ist. Nur 10 % der nicht-synonymen Tumorzellmutationen können mutierte Peptide mit hoher MHC-Affinität erzeugen, und nur 1 % der MHC-gebundenen Peptide werden von den T-Zellen der Patienten erkannt.
3. Verlust von neuen Antigenen: Das Fehlen tumorspezifischer neuer Antigene kann eine entscheidende Strategie zur Umgehung des Immunsystems von Tumoren sein. Der Verlust neuer Antigene kann auf verschiedenen Wegen herbeigeführt werden, z. B. durch Kopienzahlverlust, Transkriptionssuppression, epigenetisches Silencing und posttranslationale Mechanismen.
4. Unzureichende Produktion neuer Antigen-spezifischer T-Zellen: Das Gendetektionsprodukt Panorama 602 von Foshu Bioscience umfasst die Erkennung des Verlusts von HLA-I-Heterozygotie zur Beurteilung des Nutzens einer adjuvanten Immuntherapie. Darüber hinaus enthält es Informationen über zielgerichtete Therapien, Chemotherapie, Tumormutationslast (TMB), Tumor-Neoantigenlast (TNB), positive und negative Faktoren des Immunsystems und andere relevante Informationen über Medikamenteneinsatz, Subtypisierung, Prognose, Genetik usw., um den Patienten den größtmöglichen Nutzen zu bieten.
Referenzen
1. Nat Rev Cancer. 2017 Apr;17(4):209-222.
2.Viral Immunol. 2020 Apr;33(3):160-178.
3.Signal Transduct Target Ther. 2023 Jan 6;8(1):9.
4.Science. 2015 Apr 3;348(6230):69-74.
Statement: Dieser Artikel ist nur zur Weitergabe bestimmt. Sollte es Probleme mit dem Urheberrecht geben, kontaktieren Sie uns bitte so schnell wie möglich, und wir werden das Problem so schnell wie möglich beheben. Wir danken Ihnen!