Automatische Übersetzung anzeigen
Dies ist eine automatisch generierte Übersetzung. Wenn Sie auf den englischen Originaltext zugreifen möchten, klicken Sie hier
#Produkttrends
{{{sourceTextContent.title}}}
Pulmonale Atresie (PA)
{{{sourceTextContent.subTitle}}}
Auch bekannt als Pulmonalklappenatresie
{{{sourceTextContent.description}}}
Bei der Pulmonalklappenatresie handelt es sich um eine angeborene Herzerkrankung, die durch die Verschmelzung der Pulmonalklappensegel zu einer undurchlässigen Membranstruktur gekennzeichnet ist, wodurch der Blutfluss zwischen der rechten Herzkammer und der Lungenarterie behindert wird. Die Erkrankung wird in zwei Haupttypen eingeteilt: Pulmonalatresie mit intaktem Ventrikelseptum und Pulmonalatresie mit Ventrikelseptumdefekt. Erstere ist häufig mit einer interatrialen Kommunikation und einem offenen Ductus arteriosus (PDA) verbunden, während letztere häufig mit einer Fallot-Tetralogie-ähnlichen Anomalie einhergeht.
Die Ätiologie umfasst sowohl funktionelle Faktoren (wie pulmonale Hypertonie) als auch strukturelle Faktoren (wie rheumatische Erkrankungen und angeborene Fehlbildungen). Die Diagnose stützt sich hauptsächlich auf Echokardiographie, Ventrikeldruckmessung und Angiographie, wobei je nach Subtyp unterschiedliche Diagnosestrategien angewandt werden.
Klassifizierung der Pulmonalatresie
Die Pulmonalatresie wird in vier Haupttypen eingeteilt, die auf der Entwicklung der Pulmonalarterie, der pulmonalen Blutversorgung und dem Vorhandensein oder Fehlen eines Ventrikelseptumdefekts (VSD) basieren.
Typ I: Pulmonale Atresie mit intaktem Ventrikelseptum (PA/IVS)
- Anatomie: Vollständige Pulmonalklappenatresie mit normal ausgebildeten Haupt- und Nebenpulmonalarterien; kein VSD
- Pulmonale Blutversorgung: Abhängig von PDA oder kleineren Kollateralgefäßen
- Klinische Merkmale: Schwere Zyanose und Atemnot kurz nach der Geburt aufgrund des Duktusverschlusses
- Behandlung: Notfallmäßige Aufrechterhaltung der Duktusdurchgängigkeit (z. B. Prostaglandin E1), gefolgt von einer stufenweisen interventionellen oder chirurgischen Behandlung.
Typ II: Pulmonale Atresie mit ventrikulärem Septumdefekt (PA/VSD)
- Anatomie: Pulmonalklappenatresie kombiniert mit VSD; Pulmonalarterien sind oft hypoplastisch
- Pulmonale Blutversorgung: Hauptsächlich durch große aortopulmonale Kollateralarterien (MAPCAs) versorgt
- Assoziierte Anomalien: Häufig verbunden mit Fallot-Tetralogie-ähnlichen Merkmalen
- Herausforderungen bei der Behandlung: Erfordert eine komplexe, schrittweise chirurgische Rekonstruktion der Pulmonalarterien und der kollateralen Integration
Typ III: Fehlen der Pulmonalen Hauptschlagader
- Anatomie: Vollständiges Fehlen der Pulmonalhauptarterie; linke und rechte Pulmonalarterie entspringen direkt aus der Aorta oder den Kollateralgefäßen
- Pulmonaler Kreislauf: Völlig abhängig vom systemischen Kreislauf
- Komplikationen: Hohes Risiko für pulmonale Hypertonie und fortschreitende pulmonale Gefäßerkrankung
- Aussichten: Die chirurgische Korrektur ist sehr komplex; einige Patienten kommen nur für eine palliative Behandlung in Frage
Typ IV: Einseitige Atresie oder Fehlen der Pulmonalarterie
- Anatomie: Atresie oder Fehlen einer Lungenarterie (in der Regel der rechten), mit relativ erhaltener Entwicklung der kontralateralen Lungenarterie
- Pulmonale Blutversorgung: Die intakte Lunge unterstützt den Großteil des Lungenkreislaufs
- Behandlungsstrategie: Der Schwerpunkt liegt auf der Optimierung des Blutflusses zur funktionellen Lungenarterie; ein interventioneller Verschluss abnormaler Kollateralen kann in Betracht gezogen werden
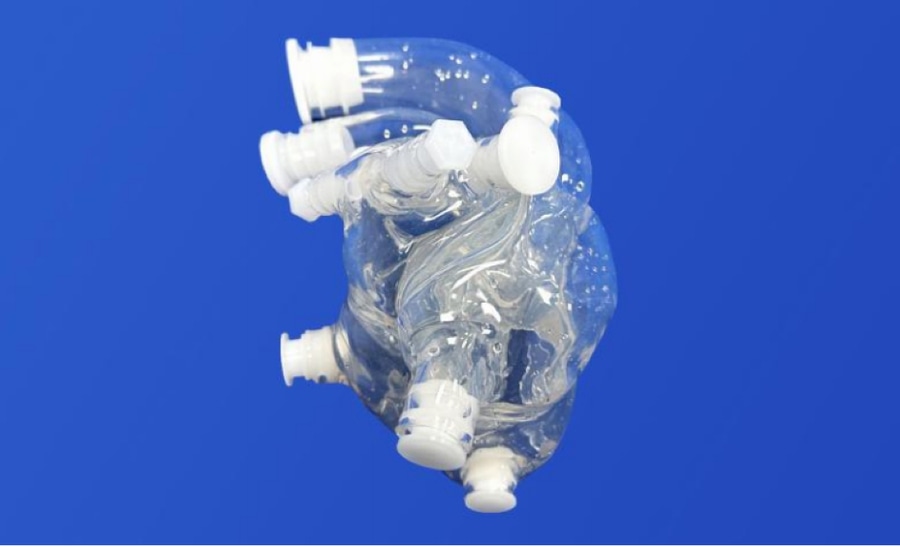
In dieses Herzmodell mit Klappen können wir eine Pulmonalklappe mit Pulmonalatresie einbauen. Unser Modell soll Ihnen helfen, die anatomischen Merkmale und die Pathophysiologie der Pulmonalatresie besser zu verstehen und die Simulation von verfahrenstechnischen und chirurgischen Behandlungsansätzen für diese Erkrankung zu unterstützen.





